Лечение фенилкетонурии
Диагностика заболевания входит в программу неонатального скрининга. Его должны пройти все без исключения новорождённые. Тест проводится на 3-5 день жизни ребёнка. Если новорождённый появился недоношенным, процесс повторяют на 7 день жизни. Исследование осуществляется с помощью забора капиллярной крови. Если обнаруживается гиперфенилаланинемии, ребенка направляют к детскому генетику. Чтобы подтвердить или опровергнуть диагноз, проводятся следующие исследования:
- выявляется активность печеночных ферментов;
- выполняется МРТ головного мозга;
- проводится биохимический анализ крови;
- Осуществляется ЭЭГ;
- выявляется концентрация тирозина и фенилаланина в крови ребенка.
Специфического лечения заболевания не существует. Основополагающим методом для борьбы с патологией выступает строгое соблюдение диеты. Она ограничивает поступление белка в организм больного. Для кормления грудного ребенка применяются специальные смеси. Они же используются и для детей старшего возраста. Основы меню становится низкобелковые продукты, которыми являются фрукты, овощи и аминокислотные смеси.
В более старшем возрасте суть диеты состоит в отказе от современных сладостей. Статистика показывает, что такие продукты содержат большое количество консервантов и эмульгаторов. Нередко в них добавляют фениланин или аспартам, оказывающие губительное влияние на больных патологией. Под запретом оказываются и:
- мясо любой природы;
- животный белок в продуктах;
- любые молочные продукты;
- растительные белковосодержащие продукты (частично);
- рыба.
Специалисты ведут большие споры, касающиеся возможности употребления материнского молока новорождёнными. Сегодня существуют две теории. В соответствии с одной из них, лучше перевести ребёнка на аминокислотные смеси, которые не содержат фенилаланин. Приверженцы другой теории настаивают, что грудное молоко необходимо для нормального роста и развития ребёнка. Оно стимулирует иммунную систему, укрепляет связь между матерью и ребенком. Однако в этом случае женщина должна сильно скорректировать свой рацион. Избирательно нужно подходить даже к витаминным комплексам. Нужно помнить, что риск обострений заболевания возрастает при малейших отступлениях от правил.
Обычно употребление вещества ребенком ограничивают до начала гормональной перестройки. В период полового созревания проблема может исчезнуть самостоятельно. Однако известны случаи, когда заместительная терапия и диета требуются пожизненно.
Если питание ребенка скорректировано до 8 недели жизни, это способно дать наибольший эффект. Если диетические ограничения были введены после 2 лет, это позволит лишь снизить выраженность симптомов.
Дополнительно может быть назначено употребление витаминов группы B, ноотропных средств, минеральных соединений и антиконвульсантов. В состав терапии в обязательном порядке должна входить лечебная физкультура, массаж и иглорефлексотерапия.
При атипичной форме заболевания диета пользы не приносит. Патология с ее помощью коррекции не поддается. В этом случае врачи назначают прием гепатопротекторов и противосудорожных средств. Такое лечение дает возможность облегчить состояние ребенка.
Дополнительно могут применяться ферментосодержащая терапия. В первую очередь врач назначает фенилаланинлиазу (PAL). Иногда принимается решение о необходимости использования кофаторама и его аналогов. Это фермент, позволяющий наладить процесс утилизации фенилаланила. База рассчитывается в соответствии с состоянием здоровья пациента, возможным прогнозом и возрастом.
История
Открытие такого заболевания, как фенилкетонурия, связывают с именем норвежского врача Ивара Асбьерна Феллинга, который в 1934 году описал гиперфенилалалинемию, связанную с задержкой в умственном развитии. В современной Норвегии патология известна также как «болезнь Феллинга».
Первые случаи успешного лечения заболевания были отмечены в Англии в начале 50 годов прошлого века. Большого прорыва в вопросе терапии добилась группа медиков под руководством Хорста Биккеля.
Но настоящего успеха в плане борьбы с фенилкетонурией удалось добиться только при разработке методов ранней диагностики болезни у новорожденных детей (метод Гатри). Это лабораторный тест, во время которого в крови определяется уровень фенилалалина. Метод был разработан и внедрен в 1958 – 1961 годах.
Со временем, в ходе многочисленных изучений и экспериментов, было установлено, что фенилкетнурия развивается при дефектах гена РАН, который является геном фенилалалингидроксилазы. Параллельно с этим были выявлены и подробно описанные атипичные формы заболевания, и разработанные современные методы терапии. Большие надежды возлагаются на проведение генотерапии в ближайшем будущем, поскольку она уже показала довольно высокие результаты в лечении некоторых наследственных заболеваний.
Фенилкетонурия: симптомы заболевания
Здесь, прежде всего, важно отметить, что первые недели жизни ребенка не позволяют внешне определить данного заболевания. Первые его признаки проявляются через два-шесть месяцев с момента рождения малыша
Он становится вялым, наблюдается отсутствие заинтересованности в отношении условий, его окружающих и мира в целом. Также ребенок становится беспокойным, нарушениям подвергается мышечный тонус.









Появляется рвота, судороги, тяжелые кожные экземы. Под экземами в частности подразумевается острая или хроническая форма воспалительного и незаразного заболевания, при котором образуется сыпь. Природа заболевания аллергическая, дополнительными проявлениями симптоматики выступает чувство жжения и выраженный зуд кожи. Актуальна и склонность к рецидивам, то есть к повторному возникновению симптоматики после относительного временного его затишья.
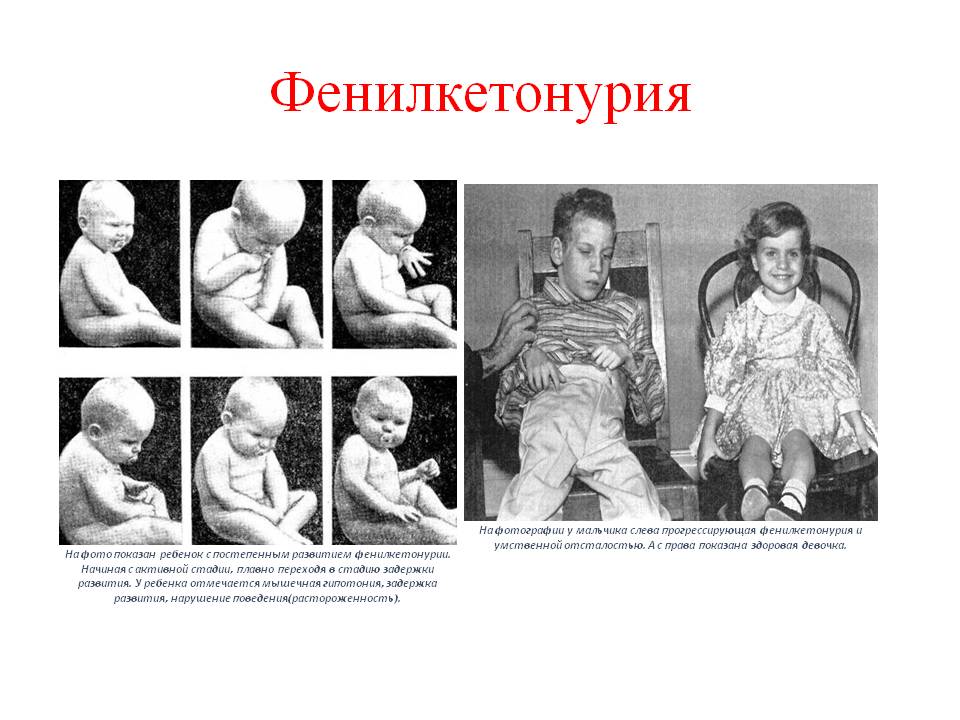
Шестой месяц позволяет определить отставание в развитии у ребенка. Одновременно с этим теряется способность к фокусированию взгляда на отдельных предметах, малыш перестает узнавать родителей. Отсутствует реакция на цветные/яркие игрушки
Важно оперативно приступить к лечению, в противном случае отсталость развития будет постепенно подвергаться лишь прогрессированию в актуальных для нее процессах

Физическое развитие больных младенцев на физическом уровне отмечается меньшими нарушениями, нежели на психическом. В обхвате голова может быть несколько меньших размеров, чем это предусмотрено для показателей нормы. Зубки прорезываются позже, позже ребенок начинает сидеть и ходить.
Принятие положения стоя у таких малышей сопряжено с широким расставлением для этого ног, а также со сгибанием их в коленях и в тазобедренных суставах, плечи и голова при этом опущены. Что касается особенностей ходьбы у больных детей, то она характеризуется покачиваниями, шажки небольшие. Сидят дети, поджав под себя ножки, что обуславливается значительным мышечным тонусом, который они испытывают.
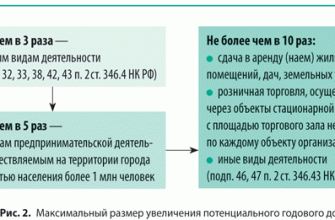
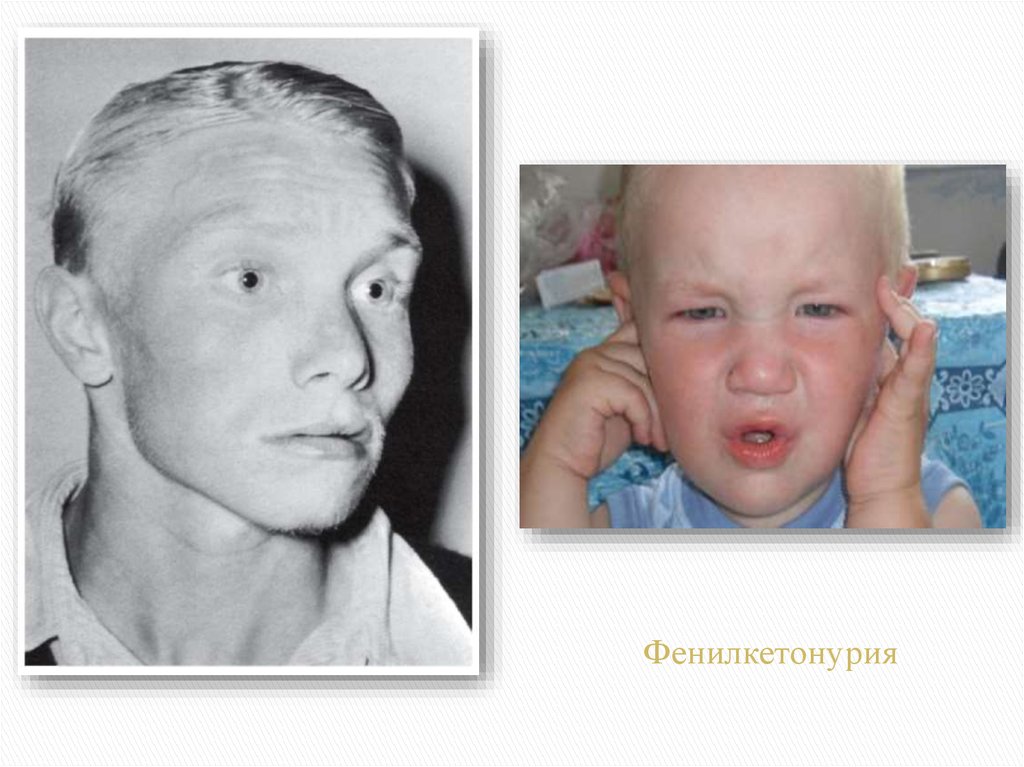
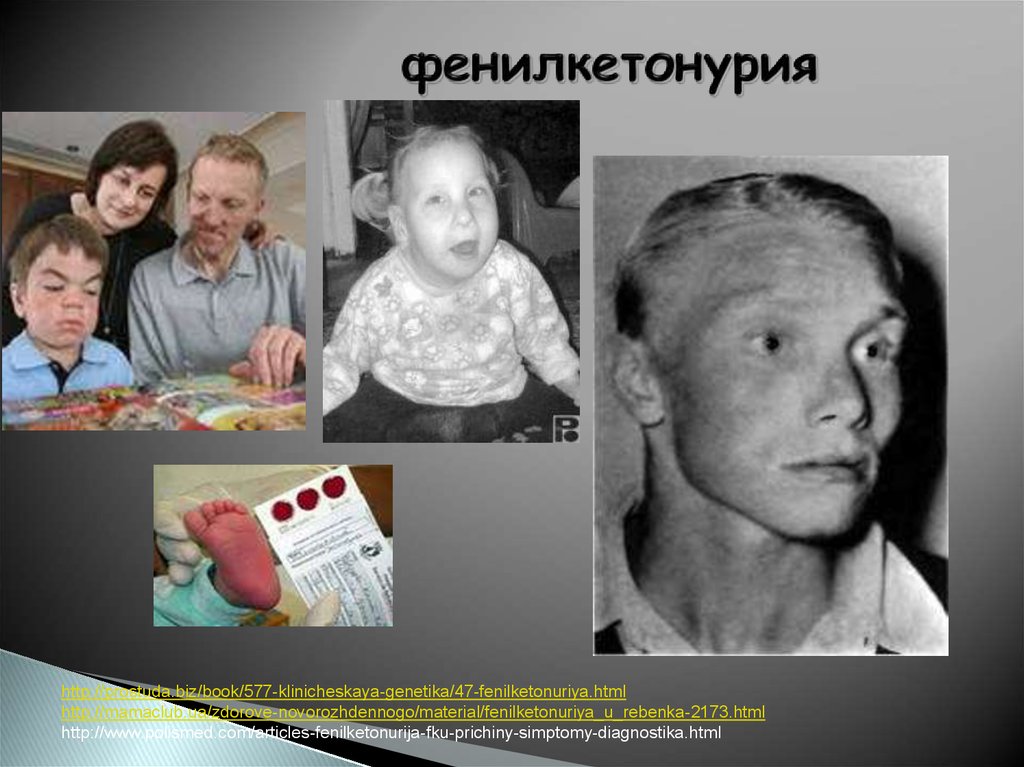
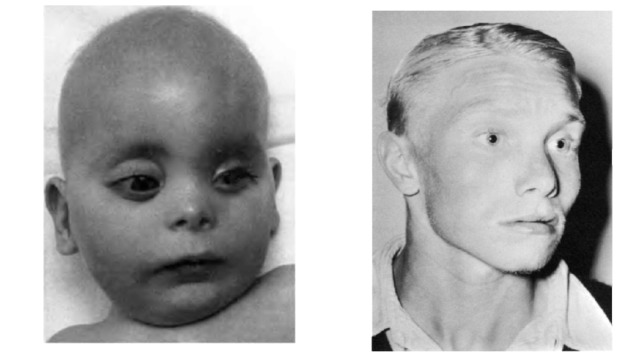
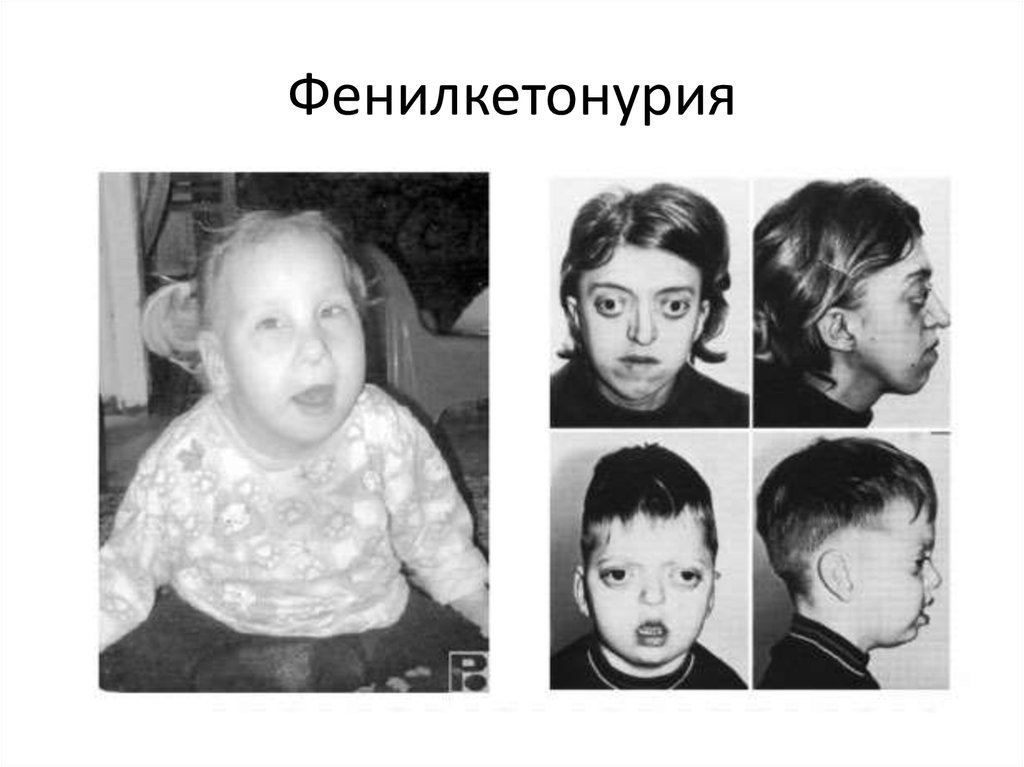
Отличаются детки и характерной внешностью со светлыми волосами, кожа у них абсолютно белая, без пигментации, глаза светлые. Учитывая излишнюю белизну кожи, она нередко покрывается у детей сыпью, что объясняется ее особой чувствительностью в отношении воздействия ультрафиолетового излучения.
В числе основных проявлений, свойственных фенилкетонурии, можно также выделить характерный «мышиный» запах, в некоторых случаях возможны эпилептические припадки, которые, однако, с возрастом исчезают. Выраженными проявлениями выступают синюшность конечностей, дермографизм (изменение в окраске кожи местного типа, происходящее при механическом воздействии на нее), потливость.
Чаще среди больных фенилкетонурией отмечается, помимо перечисленных симптомов, наличие артериальной гипотонии, дерматита, частых запоров, тремор (т.е. дрожание), потеря равновесия и расстройство в виде нарушения координации движений.
Симптомы фенилкетонурии
Симптомы заболевания у детей с классической формой патологии сразу после рождения не наблюдается. Однако такие дети обладают рядом специфических внешних признаков. У них имеет место быть:
- суховатая кожа белого оттенка;
- пигментация отсутствует практически полностью;
- волосы светлого оттенка;
- голубые глаза.
В возрасте 2 — 6 месяцев начинают проявляться симптомы заболевания. К вышеуказанным признакам добавляется вялость, пассивное восприятие окружающего мира, повышенная раздражительность и задержка психомоторного развития. Иногда возможно возникновение частой рвоты. Появляется беспричинное беспокойство. Могут наблюдаться приступы судорог. Если патологий не выявлено, в рацион ребенка вводят белковую пищу, и симптоматика начинается нарастать. Череп такого ребенка несколько уменьшен в размерах. Дети, страдающие патологией, начинают ходить позже сверстников. В год такие пациенты не могут выразить голосом эмоции. Они не воспринимают речь взрослых. Возможна задержка роста.
Не преобразованный фенилаланин выходит с потом и мочой, что приводит к появлению специфического затхлого запаха. Патология проявляется также в своеобразных позах и походке. Они возникают из-за того, что мышечный тонус больного повышен. В положении стоя ребенок широко расставляет ноги и сгибает их в коленях и тазобедренных суставах. При этом голова и плечи опущены. Походка шатающаяся. Ребёнок делает мелкие шаги. Больные сидят в так называемой позе портного. Они поджимают и скрещивают ноги. После 3 лет наблюдается повышенная возбудимость ребенка и его быстрая утомляемость. Присутствует нарушение поведения и психические расстройства. Наблюдается умственная отсталость. Очень часто болезнь сопровождает экзема, дерматит и аритмия. Если лечение отсутствует, состояние больного ухудшится. Своевременная постановка диагноза позволит уменьшить нарушения, с которыми может столкнуться ребёнок.
Что такое «фенилкетонурия»?
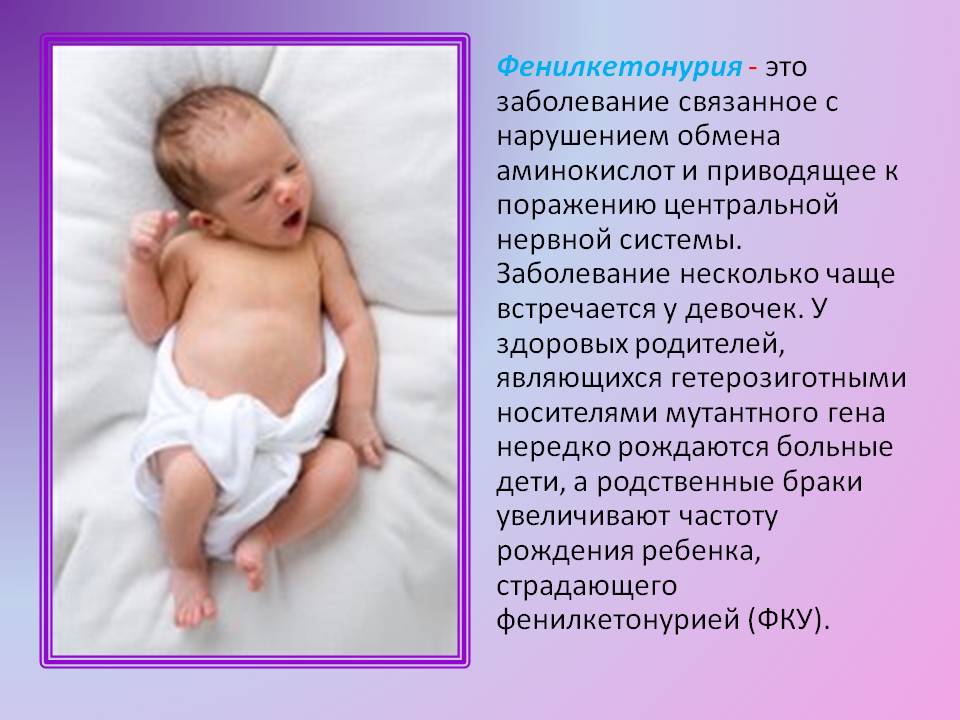
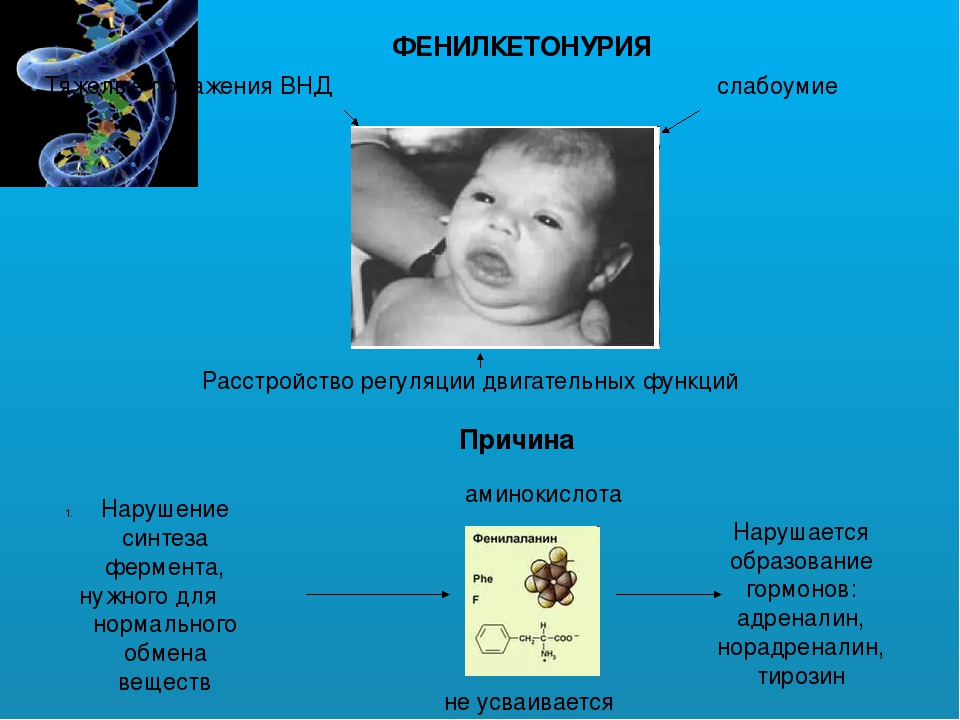
Фенилкетонурия — врожденное генетическое заболевание, которое связано с нарушением обмена аминокислот. Вещество поступает в организм человека в составе белковой пищи. В результате происходит накапливание фенилаланина и его токсичных производных. В результате наблюдается поражение ЦНС. Патология характеризуется умственной отсталостью, когнитивными и поведенческими нарушениями. У детей наблюдается явное отсутствие желания и мысли сопоставлять всё вокруг и анализировать. Возможно появление пассивного интереса, но не более. Присутствует проблема с восприятием и адаптацией к социуму. Постепенно страдает и эмоциональная сфера. Наблюдается неустойчивость поведения.








В большинстве случаев развивается классическая форма заболевания. При ней наблюдается снижение активности печеночного фермента. Он носит название фениланин-4-гидроксилаз.
Существуют и другие разновидности патологии. В 1% случаев развивается атипичная разновидность заболевания. Она появляется в результате мутации другого типа генов. Он отвечает за процесс кодирования ферментов. Наследование заболевания осуществляется в соответствии с аутосомно-рецессивным схемой.
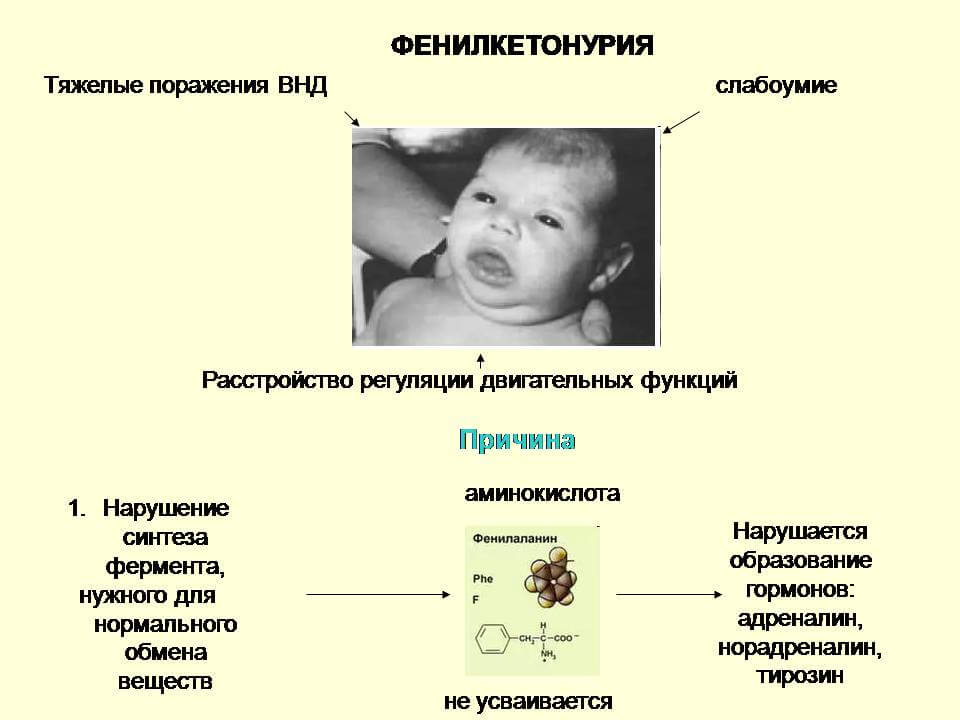
Из-за присутствия метаболического блока происходит активация побочных путей обмена фенилаланина. Это приводит к накоплению токсичных производных его действия. В частности, ими являются фениломолочная и фенилпировиноградная кислоты. При нормальном состоянии организма вещество практически не образуется. Кислоты оказывают влияние на работу ЦНС. В результате наблюдается нарушение белкового обмена, а также обмена липопротеидов и липопротеидов. Одновременно присутствуют расстройства в транспорте аминокислот. Нарушается обмен серотонина и катехоламинов. Актуальным становится перинатальные факторы.
Дополнительно начинают образовываться ортофенилацетат и фенилэтиламин. В норме они также не вырабатываются. Возможно в присутствии лишь незначительного количества осадков. Избыток приводит к нарушению метаболизма липидов. Процесс осуществляется в головном мозге. Всё это становится причиной прогрессирующей патологии интеллекта. Человек может достичь даже состояние идиотии. Окончательно процесс развития патологии пока не ясен.
Как лечить фенилкетонурию

К сожалению, лекарственных средств, которые позволили бы контролировать показатели аминокислоты без соблюдения диеты, пока нет. Атипичные формы болезни корректируются с помощью препаратов тетрагидробиоптерина и его аналогов. Для заболевания I типа единственным действенным методом лечения фенилкетонурии остается диетотерапия. Ее принцип основан на ограничении приема фенилаланина, содержащегося в продуктах. Хлеб, орехи, бобовые, рыба, мясо, творог, крупы, яйца, шоколад и др. следует исключить из рациона. Для детей первого года жизни и тем, кто немного постарше, разработаны специальные смеси, максимально приближенные к грудному молоку. Также существуют варианты для беременных и детей, достигших 6-летнего возраста. Если ограничивать поступление в организм фенилаланина с пищей до полового созревания, то патологических изменений можно избежать. Однако следует учитывать, что при отсутствии лечения резвившиеся нарушения в тканях мозга являются необратимыми, поэтому лечебная диета должна применяться с самого рождения.
Для восполнения дефицита белка, витаминов и микроэлементов, который неминуем при ограничительной диете в течение длительного времени, необходимо компенсировать недостаток специальными препаратами (белковые гидролизаты, смеси аминокислот, витамины, препараты железа и т.д.). Кроме того, в медикаментозное лечение входит прием препаратов, которые улучшают сосудистую микроциркуляцию. В зависимости от состояния здоровья назначаются антиконвульсанты.
Дети с фенилкетонурией должны находится на постоянном учете у педиатра и психоневролога. Нередко пациенты нуждаются в помощи логопеда. Поначалу контроль показателей проводится каждую неделю, затем, после нормализации состояния, ребенок до года обследуется один раз в месяц, дети более старшего возраста должны посещать врача раз в два месяца.
Какие смеси использовать для ребенка с фенилкетонурией?
Для детей с ФКУ были разработаны специальные смеси, содержащие все необходимые вещества – витамины, минералы, незаменимые и заменимые аминокислоты. Грудничкам младше годовалого возраста могут рекомендоваться следующие элементы искусственного питания:
- Афенилак 13 и 15;
- MIDмил ФКУ 0;
- ХР Аналог;
- Фенил Фри 1.
Для детей от 1 года и взрослых пациентов с ФКУ подойдут:
- П-АМ 1, 2, 3;
- Изифен;
- ХР Максамейд и Максамум с нейтральными фруктовыми вкусами.
Эти смеси имеют хорошие вкусовые качества, и легко переносятся организмом. Они особенно необходимы всем пациентам с ФКУ в периоды повышенной умственной и физической активности.
История
Фенилкетонурия как болезнь, протекающая с задержкой интеллектуального и психического развития, открыта в 1934 году норвежским ученым-исследователем Иваром Асбьером Феллингом, отсюда и другое обозначение патологии — болезнь Феллинга.
Первых успехов в коррекции здоровья малышей с ФКУ добились медики во главе с Хорстом Биккелем в середине прошлого века. Разработка терапии и ее внедрение проводились на базе Бирмингемского детского госпиталя (Англия).






Однако больших положительных результатов ученым удалось добиться, когда начала широко проводится ранняя диагностика новорожденных на выявление фенилкетонурии, это примерно 1958-1961 год прошлого века.
Расширение возможностей диагностики со временем позволило установить, что в развитии заболевания принимает участие только ген фенилаланингидроксилазы (РАН).
За последние десятилетия описаны атипичные варианты течения болезни, разработаны и широко применяются новейшие способы ее коррекции. В перспективе – использование генотерапии, что вполне вероятно позволит полностью победить болезнь.
Лечение фенилкетонурии
На сегодняшний день самым эффективным и распространенным способом лечения фенилкетонурии является элиминационная диета: диета с исключением продуктов, содержащих фенилаланин
Если ее строго придерживаться в первые годы жизни ребенка, когда развитие нервной системы еще продолжается, то можно вырастить здорового и полноценного человека. Очень важно исключение фенилаланина именно в первый год жизни, когда наиболее активно развивается нервная система. Если элиминационная диета назначается после года, умственные нарушения не излечиваются
Каждый месяц первого года жизни без применения диеты обходится ребенку безвозвратной потерей около 4 баллов IQ. Обычно достаточно придерживаться диеты до 16-18 лет, после этого возраста организм становится менее чувствительным к токсическому действию фенилаланина, и возможно расширение рациона питания. Включение новых продуктов необходимо проводить под контролем содержания фенилаланина в крови. Иногда требуется пожизненное строгое соблюдение диеты. Беременным женщинам и женщинам, планирующим беременность, и при этом больным фенилкетонурией, для рождения здорового ребенка обязательно строгое соблюдение диеты.
Степень строгости диеты зависит от концентрации фенилаланина в крови у ребенка. При его уровне до 2-6 мг% (120-360 мкмоль/л) диета не назначается, выше этого показателя – обязательна.
Суть диеты заключается в исключении белковых продуктов.
Отказ от грудного вскармливания не обязателен, но в этом случае кормящая мать должна строго придерживаться элиминационной диеты, потому что грудное молоко содержит белок (соответственно и фенилаланин). Вопрос о возможности грудного вскармливания решается индивидуально!!!
 Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
В России обеспечение лечебным питанием детей, больных фенилкетонурией, по закону бесплатное.
Больным фенилкетонурией противопоказаны следующие продукты: мясо, рыба (и морепродукты), орехи, творог, твердый сыр, бобовые, яйца, изделия из пшеничной муки, гречневая и манная крупа, овсяные хлопья.
Во время назначения элиминационной диеты необходим строгий контроль содержания фенилаланина в крови: первые 3 месяца жизни – каждую неделю, от 3-х месяцев до года – минимум раз в месяц, от года до 3-х лет – 1 раз в 2 месяца. Стремятся к содержанию фенилаланина 2-6 мг% у младших детей, после 10 лет – до 10 мг%. Обязательно наблюдение у детского психоневролога.
 Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.
Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.






Атипичные формы фенилкетонурии не поддаются лечению элиминационной диетой. В этом случае показано применение гепатопротекторов, антиконвульсантов, препаратов с Леводопой (для коррекции гиперкинезов), 5-окситриптофана, Тетрагидробиоптерина (ВН 4). Эти формы фенилкетонурии имеют худший прогноз для жизни и тем более интеллектуального развития.
На сегодняшний день разрабатываются новые направления в лечении фенилкетонурии. Среди них стоит отметить следующие:
- использование заместительной терапии фенилаланинлиазой (PAL) – растительным ферментом, расщепляющим фенилаланин до нетоксических соединений;
- генная инженерия (введение искусственно созданного нормального гена, ответственного за фенилаланин-4-гидроксилазу);
- метод «больших нейтральных аминокислот» — уменьшение всасывания фенилаланина из пищи и поступления в головной мозг с помощью специальных препаратов.
Пока эти современные разработки не имеют широкого применения, но некоторые исследования, подтверждающие их эффективность, уже проводятся.
Симптомы фенилкетонурии
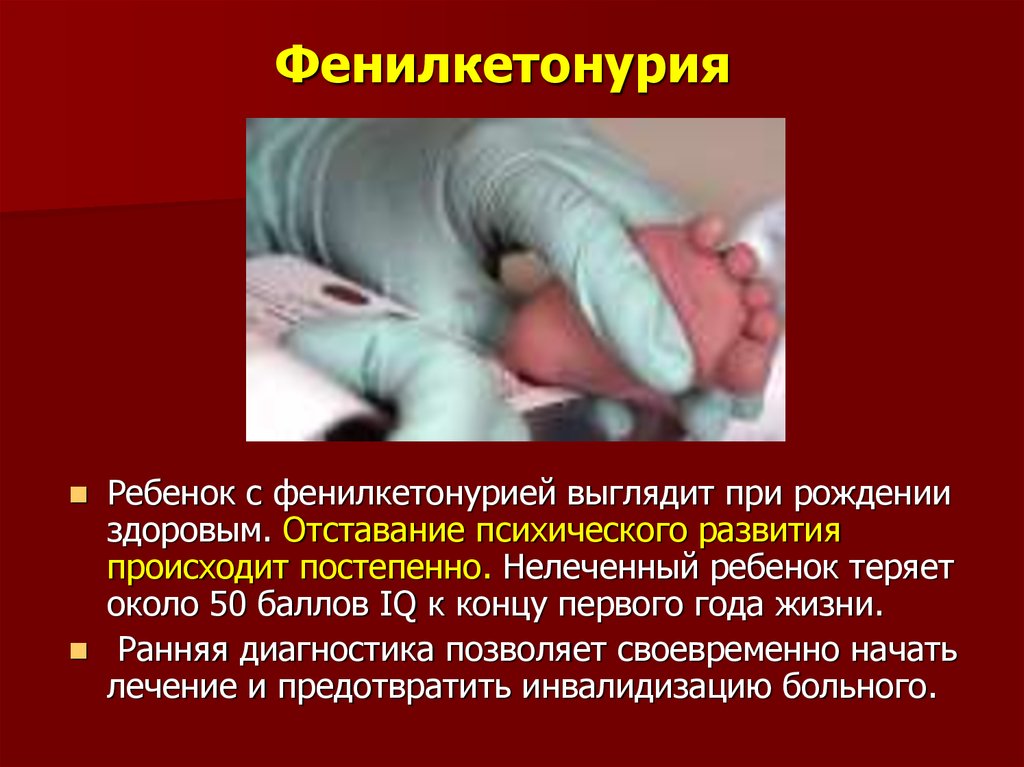
Дети с ФКУ рождаются абсолютно здоровыми. Но если болезнь была выявлена в раннем возрасте и для ее устранения были приняты все необходимые меры (назначена диета и дополнительная терапия), то дальнейшее разрушение мозга можно остановить. При этом никаких клинических проявлений патологии не возникает – ребенок растет и развивается так же, как и другие малыши его возраста.
Если же патология не была диагностирована, либо больной ребенок продолжает употреблять в пищу белковую пищу, содержащую фенилалалин, у него начинают проявляться симптомы поражения ЦНС. Для начала они выражены крайне слабо, поэтому не всегда даже опытный педиатр или невролог может их заметить. Первые признаки болезни проявляются слабостью, беспокойством, гиподинамией и апатичностью.
В полугодовалом возрасте ФКУ становится более выраженной. Малыш не проявляет особого интереса к окружающей его обстановке, не узнает родителей, не выказывает попыток сесть или перевернуться на бок. Фенилалалин и его метаболиты выводятся из организма вместе с уриной и потом. Это сопровождается появлением специфического «мышиного» или затхлого запаха.
Детки в 1 год не могут выразить вслух эмоции или переживания, их мимика скудная, невыразительная, а речь родителей для них непонятна. У деток старше 3 лет симптоматика болезни продолжает нарастать, проявляясь:
- повышенной психомоторной возбудимостью;
- быстрой утомляемостью;
- проблемным поведением;
- психотическими расстройствами;
- умственной отсталостью.
При отсутствии лечения состояния ребенка будет только ухудшаться.
Ответы на часто задаваемые вопросы
Как проявляется фенилкетонурия у новорожденных?
Как выглядят больные фенилкетонурией?
- посветление волос и радужки глаза из-за недостатка пигмента меланина
- чрезмерная прибавка в весе
- быстро зарастает большой родничок
- суховатая кожа
- шелушение, сыпь и экзема
- частая рвота
- моча и пот с характерным «мышиным» запахом
- появляются судороги и спазмы
- скованность движений и зажатая «поза портного», что связанно с повышенным напряжением в мышцах
- неадекватное поведение, выкрики, смех
- уменьшение размеров черепа
- деформация ушных раковин
- дрожание пальцев рук
- недержание мочи
- выступающая вперед нижняя челюсть
Какие смеси использовать для ребенка с фенилкетонурией?
витаминымикроэлементыДля детей до одного года рекомендуют:
- Афенилак 13, Афенилак 15 от компании «Нутритек», Россия;
- MIDмил ФКУ 0 (Hero, Испания);
- ХР Аналог («Нутриция», Голландия);
- Фенил Фри 1 («Мид Джонсон» США).
Для детей старше одного года и для взрослых:
- П-АМ 1, П-АМ 2, П-АМ 3;
- Изифен (готовый продукт), а также ХР Максамейд и ХР Максамум с нейтральным и фруктовым вкусами («Нутриция», Голландия).
Какие бывают типы фенилкетонурии?
фенилкетонурии
- Фенилкетонурия I. Классическая и наиболее распространенная форма заболевания, описанная выше в статье. Связана с мутацией гена в 12-й хромосоме, при этом нарушается образование фермента фенилаланин-4-гидроксилазы, который превращает фенилаланин в тирозин.
- Фенилкетонурия II. При этой форме заболевания нарушение происходит в 4-й хромосоме. Нарушается выработка фермента дигидроптеридинредуктазы, который также способствует превращению фенилаланина в тирозин. Заболевание наследуется так же, как и I форма: для того, чтобы родился больной ребенок, необходимо, чтобы носителями гена были оба родителя. Распространенность фенилкетонурии II – 1 случай на 100 000 новорожденных.
- Фенилкетонурия III. В результате генетических нарушений возникает недостаток фермента 6-пирувоилтетрагидроптеринсинтазы. Наследуется, как и две предыдущие формы заболевания. Распространенность – 1 случай на 300 000 новорожденных.
Дают ли инвалидность при фенилкетонурии?
Критерии установления инвалидности при фенилкетонурии
- При фенилкетонурии I инвалидность устанавливают только при необратимых нарушениях со стороны центральной нервной системы, которые приводят к неврологическим расстройствам и умственной отсталости.
- При фенилкетонурии II и III типа группу инвалидности устанавливают во всех случаях.
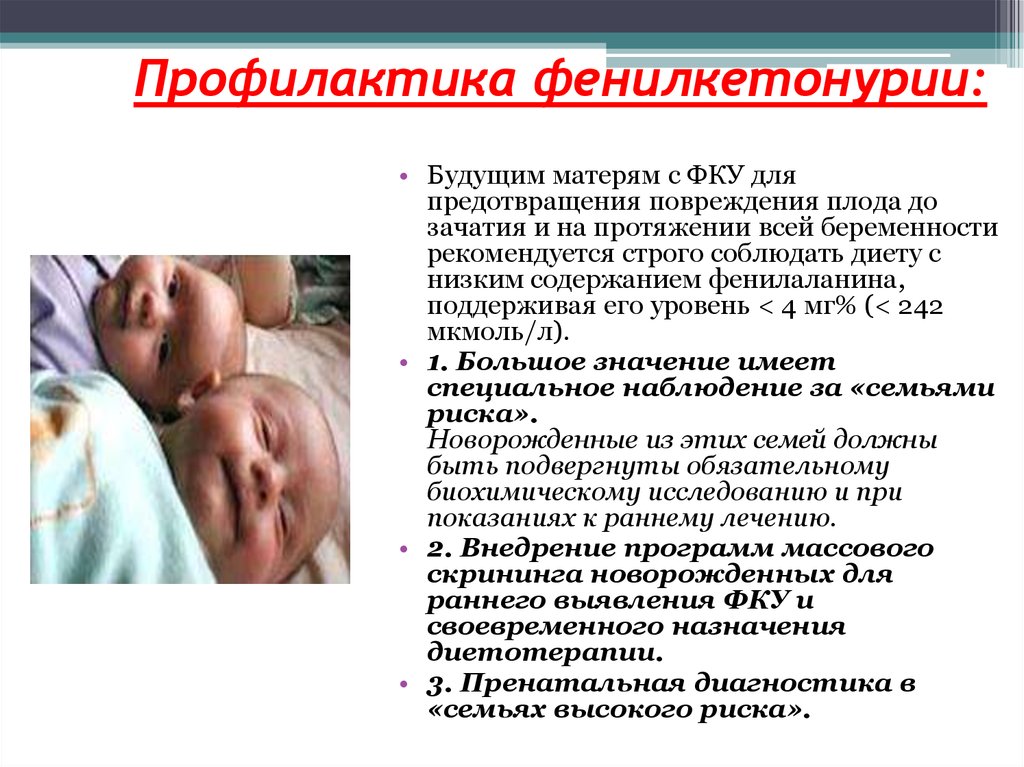
Существует ли профилактика фенилкетонурии?
- Генетическое консультирование. Необходимо людям, планирующим завести ребенка, которые больны или являются носителями неправильного гена, у которых болен хотя бы один близкий родственник или уже родился больной ребенок. Консультирование проводит врач-генетик. Он помогает разобраться, как ген, ответственный за фенилкетонурию, передавался в предыдущих поколениях, каковы риски будущего ребенка. Также генетик помогает с планированием семьи.
- Скрининг новорожденных. Анализ не помогает предотвратить заболевание, но позволяет выявить его максимально рано, пока оно еще не привело к необратимым изменениям в головном мозге.
- Консультации и диета для женщин, страдающих фенилкетонурией. Если вы женщина и страдаете ФКУ, вам следует проконсультироваться с врачом и спросить, когда лучше планировать беременность в вашем случае. Во время беременности нужно соблюдать правильную диету – это помогает предотвратить дефекты развития у ребенка.
Каковы факторы риска фенилкетонурии?
- Как уже упоминалось в статье, ребенок рискует получить заболевание или стать носителем мутантного гена, если он есть у обоих родителей.
- Среди разных этнических групп распространенность фенилкетонурии различается. Например, среди представителей негроидной расы неправильный ген встречается реже.
- В группе повышенного риска находятся дети матерей, страдающих фенилкетонурией. Если во время беременности женщина не придерживается специальной диеты, у ребенка могут возникать дефекты развития.
Фенилкетонурия – что это за болезнь?
Фенилкетонурия, или болезнь Феллинга, является тяжелой патологией, впервые описанной в 1934 году норвежским ученым Феллингом. Тогда Феллинг провел обследование нескольких детей с умственной отсталостью и выявил у них присутствие в моче фенилпирувата – продукта распада поступающей с едой аминокислоты фениланина, которая в организме больных не расщепляется. Фенилкетонурия – заболевание, связанное с нарушением обмена веществ врожденного характера, открытое одним из первых.









Фенилкетонурия – тип наследования
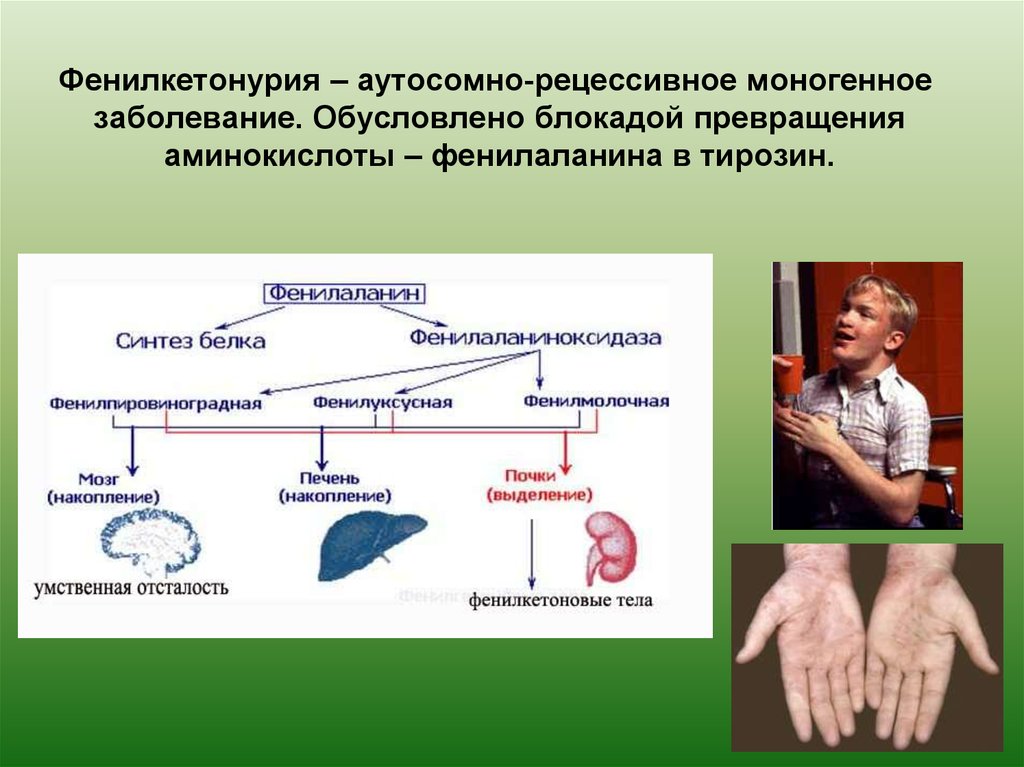
Болезнь Феллинга является хромосомно-генетической, наследственной, передаваемой детям от родителей. Виновником развития патологии выступает ген, находящийся на 12 хромосоме. Он ответственен за производство печеночного фермента фенилаланин-4-гидроксилазы, за счет которого происходит превращение фенилаланина в другое вещество – тирозин (оно требуется для нормальной работы организма).
Установлено, что фенилкетонурия наследуется как рецессивный признак. Приблизительно 2 % людей являются носителями дефектного гена, но при этом не страдают фенилкетонурией. Патология развивает только тогда, когда и мать, и отец передают ген ребенку, а случиться это может с вероятностью 25 %. Если фенилкетонурия наследуется как рецессивный признак, жена гетерозиготна, а муж гомозиготен по нормальному аллелю гена, то вероятность того, что дети будут здоровыми носителями гена фенилкетонурии, равна 50 %.
Формы фенилкетонурии
Рассматривая, у кого может развиться фенилкетонурия, что это за заболевание, зачастую речь ведется о классической форме патологии, которая встречается примерно в 98 % случаев. Остальные случаи – кофакторная фенилкетонурия, обусловленная дефектом тетрагидробиоптерина вследствие нарушения его синтеза или восстановления активной формы. Данное вещество служит кофактором ряда ферментов, и без него невозможно проявление их активности.
Фенилкетонурия – причины
Болезнь Феллинга является патологией, при которой из-за мутаций в гене, вызывающих дефицит или отсутствие фенилаланин-4-гидроксилазы, происходит накопление в тканях и физиологических жидкостях фенилаланина, а также продуктов его неполного расщепления. Часть избыточного фенилаланина превращается в фенилкетоны, выводимые с мочой, что и обусловило название болезни.
Нарушение обменных процессов сказывается в большей степени на головном мозге. На его ткани производится отравляющее воздействие, нарушаются процессы жирового обмена, происходит сбой миелинизации нервных волокон, снижение образования нейромедиаторов. Так начинается запуск патогенетических механизмов задержки умственного развития у ребенка.
Лечение
Ранее оно представляло собой ограничения, которые касались употребления фенилаланина с пищевыми продуктами (на упаковке с такими изделиями обычно находится надпись «содержит источник фенилаланина»). Но с течением времени стало понятно, что всего лишь рационального питания для лечения фенилкетонурии недостаточно.
Наилучшим вариантов является лечение, при котором уровень фенилаланина понижается до безопасного. Для этого необходимо не только контролировать питание ребенка, но и следить за его умственным развитием.
Диетотерапия на сегодняшний день является самым распространенным и эффективным способом борьбы с фенилкетонурией. Она предусматривает исключение из рациона ребенка большого количества белковой пищи – творога, мяса, яиц, бобовых, рыбы и т.д. В качестве источника жира для пациентов используют растительное или сливочное масло. Не менее важными в рационе являются фрукты, овощи и различные соки.