Причины первичного иммунодефицита
Любое заболевание, поражающее иммунную систему, изучено не в полной мере. Также обстоят дела с ПИД. Однако причины данного заболевания все же выяснены.
Генетические мутации. В этом случае изменяются гены, способствующее образованию и развитию иммунокомпетентных клеток.
Тератогенное воздействие. В утробе матери развивается плод. Токсины различного происхождения угнетают его развития, вызывая изменения на генном уровне. TORCH-инфекции часто наблюдаются при тератогенном воздействии.
Неясная этиология. Под это понятие подходят те случаи, которые не удается выявить и точно обозначить.
Вообще, даже те причины, что конкретно обусловлены медицинской наукой, имеют еще ряд серьезных вопросов. Когда код ДНК будет полностью раскрыт, то наука сможет лечить болезни без ограничений. Тогда раскроются новые горизонты. Это важный момент, ведь перспектива медицинской науки уже заложена в настоящее время.
Первичный иммунодефицит (ПИД), особенности
Является сложнейшим генетическим заболеванием, проявляющимся в первые несколько месяцев после рождения (40% случаев), в раннем младенчестве (до двух лет – 30%), в детском и юношеском возрасте (20%), реже – после 20 лет (10%).
Следует понимать, что пациенты страдают не от ИДС, а от тех инфекционных и сопутствующих патологий, которые иммунная система не в силах подавить. В связи с этим у больных может наблюдаться следующее:
- Политопный процесс. Это множественное поражение тканей и органов. Таким образом, у больного одновременно могут наблюдаться патологические изменения, к примеру, кожи и мочевыделительной системы.
- Сложность в лечении отдельно взятого заболевания. Патология часто переходит в хроническое течение с частыми рецидивами (повторениями). Болезни носят стремительный и прогрессирующий характер.
- Высокая восприимчивость ко всем инфекциям, ведущая к полиэтиологичности. Другими словами, одно заболевание может вызвать сразу несколько возбудителей.
- Обычный терапевтический курс дает не полный эффект, поэтому дозировка препарата подбирается индивидуально, часто в ударных дозах. Тем не менее, организм очень сложно очистить от возбудителя, поэтому нередко наблюдается носительство и скрытое течение болезни.
Первичный иммунодефицит является врожденным состоянием, зачатки которого образовались еще внутриутробно. К сожалению, проведение скрининга во время беременности не позволяет выявить тяжелую аномалию на первоначальном этапе.
Таблица. Причины и симптомы ПИД.
Первичный иммунодефицит
Первичный иммунодефицит является тяжелейшим наследственным генетическим нарушением (изменение в одном гене). Этот вид иммунодефицита у человека начинает проявляться практически с рождения либо в раннем детстве. Данную иммунологическую недостаточность различают в соответствии с наименованиями поврежденных компонентов (В-клетки, Т-клетки, вспомогательные клетки, фагоцитирующие клетки) или в соответствии с клиническим синдромом. Первичные иммунодефициты выявляются в 80% случаев до 20 лет.
Инфекционные процессы, которые сопровождают первичный иммунодефицит, обладают рядом отличительных особенностей:
- Политопность (множественное поражение различных тканей и органов).
- Рецидивирующее или хроническое течение болезни, склонность к прогрессированию.
- Полиэтиологичность (одновременная восприимчивость ко многим возбудителям).
- Неполный эффект лечения или неполное очищение организма больного от возбудителей.
Клиническая картина первичных иммунодефицитов (ПИД)
ПИД имеет характерный набор симптомов, которые дают возможность распознать ту или иную форму иммунной недостаточности первичного типа.
Преобладающий Т-клеточный ПИД характеризуется отставанием в физическом развитии, ранним началом, затяжными диареями, кожными высыпаниями, гепатоспленомегалией, костными аномалиями, злокачественными новообразованиями, оппортунистическими инфекциями и кандидозом полости рта.
Преобладающий В-клеточный ПИД характеризуется следующими симптомами: костно-мышечные поражения (фасцит, артрит и проч.), повторные респираторные инфекционные болезни, поражения органов желудочно-кишечного тракта, заболевания ЦНС и множество других признаков.








Дефекты фагоцитоза: заболевания мочевыводящих путей, поражение костей, поражения кожи, позднее отпадение пуповины, болезни пищеварительной системы, поражение ротовой полости, заболевания дыхательной системы, увеличение лимфатических узлов и раннее начало.
Дефекты комплемента: ревматоидные нарушения, дефицит ингибитора С1-эстеразы, повышенная восприимчивость к инфекционным процессам, первые симптомы болезни могут проявиться в любом возрасте.
Лечение иммунодефицитов
Лечение первичного иммунодефицита – довольно сложная задача. Для этого необходимо точно определить нарушенное звено иммунитета, и на основании полученных результатов назначается терапия. При недостатке иммуноглобулинов пациент нуждается в заместительной терапии на протяжении всей жизни, ему назначается сыворотка с антителами или плазма. При развитии осложнений инфекционного характера необходима антибиотикотерапия, лечение противогрибковыми препаратами и др. Иммунологическая реконструкция при первичной форме иммунодефицита возможна при пересадке костного мозга.
При вторичной форме иммунодефицита лечение также начинают с выяснения причины развития и ее устранения. Однако, в отличие от первичного иммунодефицита, терапия более эффективная. Прежде всего необходима санация очага с помощью противовирусных или антибактериальных препаратов. Терапия проводится в трех направлениях: иммунотропное лечение, заместительная терапия (плазма, иммуноглобулины, лейкоцитарная масса и др.), активная иммунизация с использованием вакцин. Вакцинотерапия может быть назначена с целью профилактики как инфекционных, так и соматических заболеваний.
Иммунодефицит у детей
Иммунодефицит – серьезный диагноз, означающий, что у малыша отсутствует естественная защита. Прикосновение к ребенку руками, вымытыми не сию минуту, родительский поцелуй и другие вполне безобидные действия с точки зрения здорового человека – источник опасности для малыша. А результат – развитие тяжелых заболеваний, при отсутствии лечения нередко приводящих к гибели.
Проблема в том, что при врожденной форме нет уникальных первичных признаков. Обычная, как считают многие родители, инфекция, желудочно-кишечные проблемы – зачастую настороженности не вызывают. А между тем болезнь приобретает хроническое течение, появляются осложнения, обычный курс антибиотиков оказывается неэффективным.
А ведь даже по характеру инфекции можно предположить о том, какой компонент иммунитета работает некорректно. Недостаточно быстрое заживление пупочной ранки, гнойное поражение кожи могут говорить о дефекте фагоцитарной системы. После полугода, как правило, появляются инфекции, связанные с исчезновением врожденного иммунитета, передаваемого от матери. Под действием патогенных возбудителей (пневмококков, стрептококков и др.) развиваются инфекции дыхательной системы. При процессах, вызываемых вирусами или грибами, можно предположить отклонения в звене Т-лимфоцитов. Тревогу должны вызвать хронические пневмонии, плохо поддающаяся терапии длительная диарея или кандидоз.
В дальнейшем характерной особенностью может быть легкость, с которой появляются и прогрессируют инфекции. К примеру, бронхит легко переходит в тяжелую пневмонию с дыхательной недостаточностью. Типичными признаками являются нарушения в работе пищеварения, папилломы, грибковые инфекции и др.
Лечение
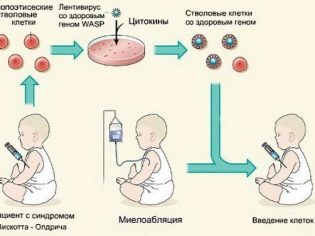
Наиболее распространённым методом лечения тяжёлого комбинированного иммунодефицита является трансплантация гемопоэтических стволовых клеток, которая проходит успешно либо при участии неродственного донора, либо при участии полу-совместимого донора, которым может являться один из родителей. Последний вид трансплантации носит название «гаплоидентичной» и был усовершенствован в Мемориальном онкологическом центре им. Слоуна-Кеттеринга в Нью-Йорке, а также в Медицинском центре дьюкского университета, где в настоящее время проведено наибольшее количество подобных пересадок. При гаплоидентичной пересадке костного мозга необходимо наличие донорского костного мозга, чтобы избежать гомологичной реакции при использовании всех зрелых T-клеток. Следовательно, функциональность иммунной системы развивается дольше у пациента, получающего костный мозг. Дэвид Веттер, один из первых, кому была проведена подобная операция, в итоге умер от вируса Эпштейна — Барр, которым был заражён костный мозг, пересаженный от его сестры. Сегодня пересадка, сделанная в первые три месяца жизни ребёнка, имеет высокий уровень успешности. Также врачи успешно проводили внутриутробную трансплантацию, сделанную до рождения ребёнка, с использованием пуповинной крови, богатой стволовыми клетками. Внутриутробная трансплантация позволяет иммунной системе плода развиваться в стерильной среде матки. Однако такое осложнение, как гомологичная болезнь, довольно сложно обнаружить.
Совсем недавно в качестве альтернативы пересадке костного мозга была предложена генотерапия. В 1990 году 4-летняя Ашанти де Сильва стала первой пациенткой, которая успешно прошла курс генной терапии. Исследователи собрали образцы крови Ашанти, изолировали некоторые лимфоциты, а затем использовали вирус, чтобы вставить в геном гены аденозиндезаминазы дикого типа. Затем эти клетки вводили обратно в организм и они начинали синтезировать нормальный фермент. Дефицит аденозиндезаминазы компенсировался дополнительными еженедельными инъекциями.
Тем не менее испытания были остановлены. В 2000 году обнаружилось, что 2 из 10 пациентов в результате генотерапии заболели лейкозом в результате введения гена, несущего ретровирус, возле онкогена. В 2007 году у 4 из 10 пациентов также был диагностирован лейкоз (данные в источнике противоречат указанной информации о лейкозе, все пациенты живы). В настоящее время работы в области генной терапии направлены на изменение вирусного вектора, чтобы уменьшить вероятность онкогенеза.
Есть также некоторые нелечебные методы терапии тяжёлого комбинированного иммунодефицита. Обратная изоляция предполагает использование ламинарного потока воздуха и механических барьеров (для избежания физического контакта с другими людьми), чтобы изолировать пациента от любых вредных патогенных микроорганизмов, присутствующих во внешней среде.
Первичные иммунодефициты[править | править код]
Определение и классификация
Первичные иммунодефициты — это врожденные (генетические или эмбриопатии) дефекты иммунной системы. В зависимости от уровня нарушений и локализации дефекта они бывают:
- гуморальные или антительные — с преимущественным поражением системы В-лимфоцитов
- Х-сцепленная агаммаглобулинемия (болезнь Брутона)
- Гипер-IgM синдром
- Х-сцепленная
- аутосомно-рецессивная
- делеция генов тяжелых цепей иммуноглобулинов
- дефицит k-цепей
- селективный дефицит субклассов IgG с или без дефицита IgA
- дефицит антител с нормальным уровнем иммуноглобулинов
- общая вариабельная иммунная недостаточность
- дефицит IgA
- клеточные
- синдром Ди Джоржи
- первичный дефицит CD4 клеток
- дефицит CD7 Т-клеток
- дефицит ИЛ-2
- множественная недостаточность цитокинов
- дефект передачи сигнала
- комбинированные:
- синдром Вискотта-Олдрича
- атаксия-телеангиоэктазия (синдром Луи-Бар)
- тяжелая комбинированная иммунная недостаточность
- Х-сцепленная с полом
- аутосомно-рециссивная
- дефицит аденозиндезаминазы
- дефицит пуриннуклеозидфосфорилазы
- дефицит молекул II класса МНС (синдром лысых лимфоцитов)
- ретикулярная дизгенезия
- дефицит CD3γ или CD3ε
- дефицит СD8 лимфоцитов
- недостаточность системы комплемента
- дефекты фагоцитоза
- наследственные нейтропении
- инфантильный летальный агранулоцитоз (болезнь Костмана)
- циклическая нейтропения
- семейная доброкачественная нейтропения
- дефекты фагоцитарной функции
- хроническая гранулематозная болезнь
- Х-сцепленная
- аутосомно-рециссивная
- дефицит адгезии лимфоцитов I типа
- дефицит адгезии лейкоцитов 2 типа
- дефицит глюкозо-6-дегидроегназы нейтрофилов
- дефицит миелопероксидазы
- дефицит вторичных гранул
- синдром Швахмана
- наследственные нейтропении
Клиническая картина иммунодефицитных состояний (ИДС)
Клиника имеет ряд общих черт:
- 1. Рецидивирующие и хронические инфекции верхних дыхательных путей, придаточных пазух, кожи, слизистых оболочек, желудочно-кишечного тракта, часто вызываемые оппортунистическими бактериями, простейшими, грибами, имеющие тенденцию к генерализации, септицемии и торпидные к обычной терапии.
- 2. Гематологические дефициты: лейкоцитопении, тромбоцитопении, анемии (гемолитические и мегалобластические).
- 3. Аутоиммунные расстройства: СКВ-подобный синдром, артриты, системная склеродермия, хронический активный гепатит, тиреоидит.
- 4. Нередко иммунодефицитные состояния (ИДС) сочетается с аллергическими реакциями 1 типа в виде экземы, отека Квинке, аллергическими реакциями на введение лекарственных препаратов, иммуноглобулина, крови.
- 5.Опухоли и лимфопролиферативные заболевания при ИДС встречаются в 1000 раз чаще, чем без ИДС.
- 6. У больных с ИДС часто отмечаются расстройства пищеварения, диарейный синдром и синдром мальабсорбции.
- 7. Больные с ИДС отличаются необычными реакциями на вакцинацию, а применение у них живых вакцин опасно развитием сепсиса.
- 8. Первичные ИДС часто сочетаются с пороками развития, прежде всего, с гипоплазией клеточных элементов хряща и волос. Кардиоваскулярные пороки описаны, главным образом, при синдроме Ди-Джоржи.
Лечение первичных ИДС
Этиотропная терапия заключается в коррекции генетического дефекта методами генной инженерии. Но такой подход является экспериментальным. Основные усилия при установленном первичном ИДС направлены на:






- профилактику инфекций
- заместительную коррекцию дефектного звена иммунной системы в виде трансплантации костного мозга, замещения иммуноглобулинов, переливания нейтрофилов.
- заместительную терапию ферментами
- терапию цитокинами
- витаминотерапию
- лечение сопутствующих инфекций
- генная терапия
- иммуномодулирующая терапия
В 2018 году российский препарат на основе высокоочищенных иммуноглобулинов класса G прошел I фазу клинических исследований. В ходе испытаний была подтверждена безопасность применения лекарственного средства. Планировалось, что после регистрации и завершения дополнительных исследований, препарат возможно будет применять в качестве заместительной и иммуномодулирующей терапии у пациентов со сниженным или отсутствующим уровнем синтеза антител. Средство направлено на обеспечение нормализации уровня иммуноглоублина до оптимальных значений и повышение сопротивляемости организма к патогенам.
Первичный иммунодефицит — статистика
Тяжелые генетические дефекты иммунной системы, порой несовместимые с жизнью, встречаются довольно редко, примерно 1 случай на 10 000 новорожденных. Однако распространенность легких форм первичных иммунодефицитов, например, селективного дефицита иммуноглобулина А, составляет 1 случай на 500-1000 человек в зависимости от расы. Согласно последним данным в мире предположительно около 6 миллионов людей страдают различными формами наследственных нарушений иммунитета. Большое число случаев первичного иммунодефицита до сих пор остается недиагностированным.
С одной стороны, цифры статистики указывают на редкость тяжелых врожденных иммунодефицитных состояний, а с другой — на вполне реальную возможность родителей столкнуться с первичным иммунодефицитом легкого течения у их малыша. Наиболее частым является нарушение выработки антител или гуморальный иммунодефицит. Он может дебютировать у детей, начиная с возраста 5 — 7 месяцев жизни, как только снижается уровень защитных материнских антител. Такое заболевание имеет достаточно благоприятное течение и хороший прогноз.
Диагностика и лечение
Диагностика и лечение состояний дефицита иммунитета у детей проводится врачами-педиатрами, иммунологами. Обычно первым догадываться о состоянии начинает именно педиатр, к которому родители малыша обращаются слишком часто по поводу воспалений, вирусных и иных недугов у чада. В этом случае доктор рекомендует сделать анализы и посетить иммунолога.
Общий осмотр включает оценку состояния кожных покровов, полости рта ребенка, наличия или отсутствия отеков. Лабораторная диагностика дает максимум информации о состоянии иммунной защиты: в общем анализе крови у ребенка нарушена лейкоцитарная формула, есть многочисленные аномалии. Биохимический анализ крови показывает наличие необычных метаболитов.
Специальные иммунологические анализы позволяют выявить активированные лейкоциты, фагоциты, количество иммуноглобулинов, селективную нехватку иммуноглобулинов. При подозрении на первичный генетический дефицит назначают молекулярно-генетический анализ, который позволяет установить факт мутаций определенных генов.

Лечение зависит от формы иммунодефицита.
Первичный. Нередко медицина не может предложить ничего существенного для избавления от грубых форм врожденного иммунодефицита. Больные умирают от осложнений инфекционных болезней. В некоторых случаях справиться с проблемой частично помогает трансплантация костного мозга. Больным с нехваткой факторов иммуноглобулинов вводят такие препараты искусственно, и терапия носит пожизненный характер. Детям зачастую противопоказана вакцинация. Прививки не только опасны, но и совершенно бесполезны — введение вакцины не формирует должного иммунитета против заболевания, от которого ее делают.

Нарушения обмена веществ могут потребовать гормональной терапии. Опухоли нуждаются зачастую в хирургическом лечении. Для детей с таким иммунодефицитом рекомендуется делать прививки активной вакциной
Это очень важно для восстановления нормального иммунного статуса. Могут проводить заменные переливания крови и донорской плазмы


Симптомы
Симптомы иммунодефицита у детей разнообразны, и могут включать не только иммунологические нарушения, но и нарушения развития, недостаточную обучаемость, расстройства сна, а также опухолевые процессы и др.
Манифестация первичного у детей наблюдается, как правило, еще в раннем возрасте. При синдроме Вискотта, например, характерна триада — повышенная кровоточивость, кожная экзема, частые рецидивы инфекций.










Характерно, что можно выделить некие общие проявления, которые схожи у заболеваний разных групп. Они могут указывать, какой компонент иммунной системы был поражен либо в каком звене или механизме имеются нарушения:
| Первичные дефициты клеточного иммунитета | Ослабление гуморальной защиты<,/th>, | Комбинированные иммунодефициты |
|---|---|---|
| В таких случаях наиболее характерны вирусные и грибковые поражения. Дети часто простывают, и вирусные инфекции имеют склонность протекать более тяжело. Сюда относят ветряную оспу, например. Герпетические поражения выраженные. Также у детей часто выявляют кандидозы — поражается полость рта, легкие, желудочно-кишечный тракт (синдром раздраженного кишечника), половые органы. При нарушениях в клеточном звене иммунной защиты увеличивается вероятность развития злокачественных образований | Характерна частая заболеваемость бактериальными инфекциями. Может наблюдаться пиодермия, пневмония, и эти заболевания отличаются тяжелым характером течения. Может быть рожистое воспаление. Часто поражаются слизистые оболочки — ротовая и носовая полости, конъюнктива. | Характеризуются и вирусными, и бактериальными поражениями. В таких случаях отмечаются не столько проявления иммунной недостаточности, сколько более специфические проявления — это может быть поражение тимуса, лимфоидной ткани, анемия, пороки развития. |
При врожденных нейтропениях характерна склонность к формированию абсцессов, которые при отсутствии своевременного и адекватного лечения приводят к флегмоне и развитию сепсиса.
Для комплемент-ассоциируемых иммунодефицитов характерно снижение устойчивости к бактериальным агентам или же развитие аутоиммунных заболеваний. Отдельно выделяют наследственный АНО, который проявляется себя периодически возникающей отечностью в разных частях тела.
Причины иммунодефицита
По этиологии различают первичные и вторичные иммунодефициты.
Первичные иммунодефициты развиваются на фоне генетических нарушений. При этом происходит нарушение отдельных составляющих иммунитета:
Гуморального ответа:
Болезнь Брутона;
- Общий вариабельный иммунодефицит;
- Селективный дефицит иммуноглобулинов;
- Транзиторная гипогаммаглобулинемия у детей.
Клеточного звена:
- Хронический слизисто-кожный кандидоз;
- Синдром Ди Джорджи.
Системы фагоцитов:
- Синдром Чедиака – Стейнбринка – Хигаси;
- Хронический гранулематоз;
- Синдром Джоба;
- Дефицит экспрессии молекул адгезии.
Комплимента: врожденный ангионевротический отек.
Выделяют также комбинированные иммунодефициты:
- Тяжелый комбинированный иммунодефицит;
- Синдром Луи-Бар;
- Комбинированный иммунодефицит с повышенным иммуноглобулином М;
- Иммунодефицит с карликовостью;
- Синдром Вискотта-Олдрича.
Первичный иммунодефицит сопровождает человека в течение всей жизни. Умирают такие пациенты от инфекционных осложнений.
Вторичный иммунодефицит развивается по причине воздействия на организм различных инфекций и неблагоприятных факторов внешней среды. Вторичные иммунодефициты (кроме вируса иммунодефицита человека) хорошо поддаются лечению и являются обратимыми.
В качестве основных причин вторичных иммунодефицитов выступают:
- Хронические вирусные и бактериальные инфекции, паразитарные инвазии (вирус иммунодефицита человека, стафилококкоз, туберкулез, пневмококкоз, токсоплазмоз, аскаридоз, малярия, лейшманиоз и другие). При инфекционных хронических заболеваниях иммунная система сильно изменяется, происходит интоксикация организма, угнетается функция кроветворения. При наличии вируса иммунодефицита человека иммунодефицит опосредуется поражением клеток иммунной системы;
- Нарушение питания и истощение организма. Иммунитет человека особенно чувствителен к недостатку минералов, витаминов, питательных веществ. Поэтому снижение защитных сил организма чаще наблюдается в период сезонной витаминной недостаточности;
- Диарейный синдром;
- Гельминтозы;
- Тяжелые травмы и операции. Любое серьёзное заболевание или оперативное вмешательство ведет к вторичному иммунодефициту. Это связано с интоксикацией организма, нарушением метаболизма, а также производством организмом большого количества гормонов надпочечников, угнетающих функцию иммунной системы;
- Стресс-синдром;
- Большие потери крови, ожоги, заболевания почек;
- Эндокринопатии (гипертиреоз, гипотиреоз, сахарный диабет);
- Острые и хронические отравления токсическими веществами, наркотическими средствами, лекарственными препаратами. Сильно снижают иммунитет цитостатики, антибиотики, глюкокортикоиды, антиметаболиты;
- Низкая масса тела при рождении;
- Злокачественные новообразования;
- Аутоиммунные заболевания;
- Старческий и детский возраст, беременность. Снижение иммунитета в этих случаях обусловленофизиологическими особенностями организма в эти периоды жизни человека.
Примечания
- Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 1-4160-2999-0
- a b «News From Indian Country — A rare and once-baffling disease forces Navajo parents to cope». Retrieved 2008-03-01
- «Wisconsin First State in Nation to Screen All Newborns for Severe Combined Immune Deficiency (SCID) or „Bubble Boy Disease“»
- «NEWBORN SCREENING FOR PRIMARY IMMUNODEFICIENCY DISEASE»
- «MDCH Adds Severe Combined Immunodeficiency (SCID) to Newborn Screening»
- «Severe Combined Immunodeficiency (SCID): Immunodeficiency Disorders: Merck Manual Professional». Retrieved 2008-03-01
- a b Chinen J, Buckley RH (2010). «Transplantation immunology: solid organ and bone marrow». J. Allergy Clin. Immunol. 125 (2 Suppl 2): S324-35
- Vickers, Peter S. (2009). Severe combined immune deficiency: early hospitalisation and isolation. Hoboken NJ: John Wiley & Sons, 29-47. ISBN 978-0-470-74557-1
- Buckley RH (2004). «Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution». Annu. Rev. Immunol. 22 (1): 625—655
- a b Fischer A, Hacein-Bey S, Cavazzana-Calvo M (2002). «Gene therapy of severe combined immunodeficiencies». Nat Rev Immunol 2 (8): 615—621
- Press release from the European Society of Gene Therapy
- Cavazzana-Calvo M, Fischer A (2007). «Gene therapy for severe combined immunodeficiency: are we there yet?». J. Clin. Invest. 117 (6): 1456—1465
- Tamaroff MH, Nir Y, Straker N (1986). «Children reared in a reverse isolation environment: effects on cognitive and emotional development». J. Autism Dev. Disord. 16 (4): 415—424
Причины
Первичные иммунодефициты обычно связаны с низкой активностью или дефицитом Т-лимфоцитов. Причины такого поведения иммунных клеток могут крыться в нарушении функций тимуса, нарушении выработки ферментов. Выделяют несколько заболеваний в этой группе. Гуморальные связаны с нехваткой В-лимфоцитов, нарушением выработки иммуноглобулина (синдром Брутона, Веста). Комбинированные первичные нарушения иммунитета связаны с низкой активностью иммунных клеток и гуморальных звеньев (синдром Гланцмана-Риникера, болезнь Луи-Бар).








Фагоцитарные первичные состояния связаны с низкой активностью моноцитов и гранулоцитов. Сюда относят так называемый синдром «ленивых лейкоцитов» и нейтропению Костмана. А белковые первичные дефицитные состояния связаны с мутацией генов, которые кодируют процессы выработки тех или иных белков, необходимых для работы иммунитета.
Вести к выраженному и стойкому снижению иммунной защиты организма могут самые разнообразные этиологические факторы – как внешние, так и внутренние.


Вторичный иммунодефицит нередко развивается при общем истощении организма. Длительное недоедание с дефицитом в рационе белка, жирных кислот, витаминов и микроэлементов, нарушения всасывания и расщепления питательных веществ в пищеварительном тракте приводят к нарушению процессов созревания лимфоцитов и снижают сопротивляемость организма.
Причины вторичных дефицитных состояний иммунитета у детей многочисленны. Спровоцировать патологические снижение естественной защиты организма могут патологии опорно-двигательной системы и внутренних органов, ожоги, большая кровопотеря, с чем бы она ни была связана.
Хронические вирусные болезни (ВИЧ, цитомегалия, краснуха, гепатит) разрушают звенья иммунитета как на клеточном, так и на гуморальном уровне. Менее разрушительны, но не менее опасны и грибковые, и бактериальные, а паразитарные заболевания.
При болезни почек, печени, эндокринных заболеваниях снижается активность фагоцитов, что также приводит к иммунодефицитному состоянию.
Если ребенок долгое время принимает лекарственные средства, влияющие на состояние костного мозга и процессы выработки клеток крови (цитостатики, глюкокортикоидные средства), снижается активность лимфоцитов. Почти таким же образом действует и радиация.
Если у ребенка есть злокачественная опухоль, то она самостоятельно способна продуцировать цитокоины, что снижает количество Т-лимфоцитов и угнетает активность фагоцитов.


Лечение
Иммунодефицитные состояния у маленьких детей требуют обязательного лечения. Вследствие различия в этиологических факторах и патогенезе различных форм первичных иммунодефицитов не существует универсального подхода в лечении.
В тяжелых случаях высок риск летального исхода в связи с инфекционными осложнениями, и терапевтические меры имеют лишь временных характер.
В некоторых случаях целесообразно проведение трансплантации костного мозга либо эмбриональной ткани тимуса.
Что можете сделать вы
При первых подозрениях как можно скорее обращаться за квалифицированной медицинской помощью.Необходимо отменить все вакцинации.
Что делает врач

На приеме у врача необходимо подробно изложить жалобы, отвечать на все вопросы и ничего не утаивать. Также педиатр уточнит о наличии родственников у детей с иммунодефицитными состояниями. Помимо детального осмотра необходимы данные лабораторных исследований. До получения результатов назначают симптоматическое лечение детей, а после постановки окончательного диагноза уже выстраивается дальнейшая стратегия и осуществляется лечение детей согласно клиническим рекомендациям.







Лекарства
При первичном гуморальном иммунодефиците требуется заместительная терапия -— вводят иммуноглобулины. Очередная инфекция, будь то бактериальная или вирусная, или грибковая, требует также лечения. Детям могут назначать повышенные дозировки, однако детей с иммунодефицитом недопустимо.
Вторичные иммунодефициты
ВИД представляют собой осложнения многих состояний и болезней. Заболеть вторичным иммунодефицитом человек может по таким причинам:
- Общее истощение организма и неправильное питание.
- Хронические вирусные и бактериальные инфекционные процессы, а также, паразитарные инвазии тоже могут стать провокаторами вторичных иммунодефицитов.
- Гельминтозы.
- Диарейный синдром.
- Сильная потеря крови (при заболеваниях почек, ожогах и проч.), которая ведет к потере факторов иммунной защиты и вторичному иммунодефициту.
- Стресс-синдром.
- Низкая масса тела в момент рождения.
- Хронические и острые отравления ксенобиотиками.
- Эндокринопатии.
- Операции и тяжелые травмы.
- Злокачественные новообразования.
- Аутоиммунные заболевания.
- Низкий иммунитет у беременных женщин и людей пожилого возраста.
Проявления вторичных иммунодефицитов
Человек с иммунодефицитом вторичного типа страдает, в основном, следующими синдромами и заболеваниями: персистирующая, тяжелая, рецидивирующая бактериальная инфекция; инфекционные заболевания слизистых оболочек и кожи; рецидивирующие респираторные инфекции; неврологические проблемы (аутоиммунные состояния, энцефалит, судорожные припадки); повышенная частота рака желудка и заболеваний печени; гематологические нарушения (тромбоцитопения, лейкопения, аутоиммунная гемолитическая анемия); расстройства желудочно-кишечного тракта (даже диарея); легкое развитие и прогрессирование осложнений (например, обычный острый бронхит за максимально короткое время может перерасти в пневмонию, бронхоэктазию и дыхательную недостаточность).
Типы
| Тип | Описание |
|---|---|
| X-сцепленный тяжёлый иммунодефицит | Наиболее распространённый тип тяжёлого комбинированного иммунодефицита, возникающий из-за мутаций в гене, кодирующем общие гамма-цепи, белок которых является общим для рецепторов интерлейкинов IL-2, IL-4, IL-7, IL-9, IL-15 и IL-21. Перечисленные интерлейкины и их рецепторы вовлечены в процессы развития T- и B-лимфоцитов. В результате мутаций происходят дисфункции общей гамма-цепи, и, как следствие, дефект распространяется на процесс сигнализации интерлейкина. Происходит почти полный отказ иммунной системы как со стороны развития, так и со стороны функционирования, с отсутствием или очень малым количеством T-лимфоцитов, NK-клеток и нефункциональными B-лимфоцитами.
Общая гамма-цепь кодируется геном IL-2 рецепторов гамма, который находится на X-хромосоме. Наследуется как рецессивный признак. |
| Дефицит аденозиндеаминазы | Второй по распространённости тип тяжёлого комбинированного иммунодефицита. Его причиной является дефект фермента аденозиндеамиазы, который необходим для расщепления пуринов. Недостаток аденозиндеаминазы провоцирует накопление dATP. Этот метаболит ингибирует активность фермента рибонуклеотидредуктазы, участвующего в превращении рибонуклеотидов в дезоксирибонуклеотиды. Эффективность иммунной системы зависит от пролиферации лимфоцитов и, следовательно, синтеза dNTP. Если рибонуклеотидредуктаза не способна нормально функционировать, пролиферация лимфоцитов блокируется, а иммунная система компрометируется. |
| Синдром Оменна | Производство иммуноглобулинов требует участия рекомбинантного фермента, полученногог от рекомбинации генов, активирующих RAG-1 и RAG-2.
Эти ферменты участвуют в первом этапе V(D)J рекомбинации, в котором сегменты B-лимфоцитов или ДНК T-лимфоцитов перестраиваются, создавая новые T- или B-клеточный рецепторы. Некоторые мутации RAG-1 или RAG-2 продотвращают процесс V(D)J рекомбинации, тем самым приводя к возникновения ТКТД. |
| Синдром голых лимфоцитов | MHC класса II не экспрессируется на поверхности антигенпредставляющих клеток. Аутосомно-рецессивный тип наследования. |
| Дефицит JAK3 | JAK3 является ферментом, который выступает посредником трансдукции через общую гамма-цепь. Мутация гена JAK3 также вызывает тяжёлый комбинированный иммунодефицит. |
| Дефицит DCLRE1C/Artemis | Несмотря на то, что исследователями было идентифицировано около дюжины генов, вызывающих ТКИД, население Навахо и Апачи страдает наиболее тяжёлой формой заболевания. Это связано с отсутствием гена DCLRE1C/Artemis. Без этого гена организм ребёнка не в состоянии восстановить ДНК или вырабатывать антитела. |